In this Surface Science educational series in two parts, you will be introduced to how it all began
The series cover topics such as the bridging of surface physics and surface chemistry, the paradigm shift from the study of static surface systems to the exploration of dynamic ones, as well as the importance of the advances in theory without which the evolution of the field could not have happened. It will also guide you through the different analytical methods that are used in the surface science
This post about the dynamics of atom/molecule interactions with surfaces, is a natural extension of the previous post, part 1, which addressed static properties of surface systems. Static systems refer to systems not changing in time, while dynamic systems refer to systems where temporal changes are an inherent characteristic. In contrast to kinetics, which

The presentation describes central concepts in molecule surface dynamics such as surface scattering, potential energy surfaces(PESs), energy exchange and transfer between a surface and an incident/desorbing molecule. It talks about surface corrugation, charge transfer and adiabatic versus non-adiabatic descriptions of the interactions. Specific surface-molecule systems are not covered, except for some examples that illustrate general concepts and types of model systems. Experimental methods are only briefly described and will be covered in later posts.
In the early development of surface science and particularly in surface physics, the focus was on the “static properties” of simple surfaces. It was challenging enough to describe the surface, with or without adsorbates, in a “frozen” situation, without involving temporal changes and dynamics.
For many years, the determination of structural aspects of adsorbed atoms and molecules, both as single adsorbates and as dense monolayers, and the associated electron structure, was a primary field. Bond positions of atoms, bond lengths, and vibrational properties were in focus together with the associated electron structure aspects, such as bond orbitals, surface band structure and charge distributions. This development was described in the first post in this series, The evolution of surface science, part 1, and more will follow in a later post, where theoretical aspects will be described.
Today the position(s), orientation(s) and electron structure of simple adsorbates, like, e.g., CO molecules, on single crystal metal surface like Ni, Cu, Pt can be described in atomistic detail and with chemical accuracy in the energies.
While the static approach addresses the frozen state, the dynamics concern the time evolution of reversible or irreversible changes of the surface system. Examples are adsorption processes and diffusion, reaction, and desorption of adsorbates. Theoretically, static systems can be described by solving the time-independent Schrödinger equation, while dynamic systems require the even more challenging task to solve the time-dependent equation.
Historically gas surface dynamics was “born” from two “parents”; molecular collision dynamics in the gas phase, an active field already during the first part of the 20th century, and the surface community, until then focusing on static surface systems (Part 1 in this blog post series). Both communities were in the 2nd half of the 20th century looking for new areas, and challenges for their studies and they merged their knowledge, techniques, and concepts in gas surface dynamics. The molecule-molecule collision community had performed their studies by using one or two molecular beams to allow gas molecules to collide under well-controlled collisions in a vacuum, with prescribed translational and internal (electronic, vibrational and rotational) energies. This community was the main contributor of the molecular beam technique and post-scattering analytical tools, while the surface community’s main contribution was the surface knowledge, e.g., how to prepare and analyze surfaces and construct potential energy surfaces for molecule-surface systems.
One strong driver towards dynamics studies was the rich, purely basic research questions in this field; to just ask and learn how a molecule scatters against a surface, Fig. 1, and is caught or reflected by it. However, a major motivation was also the relevance for technically important surface processes and reactions like heterogeneous catalysis, dry etching, CVD (chemical vapor deposition) and oxidation. Knowledge about the static systems certainly provides significant insight also about dynamic systems, but it is insufficient to develop detailed and well-founded models and scenarios for systems developing in time. It is like comparing a few photographic snapshots (the static properties) versus a movie (dynamics) to describe a system developing in time.
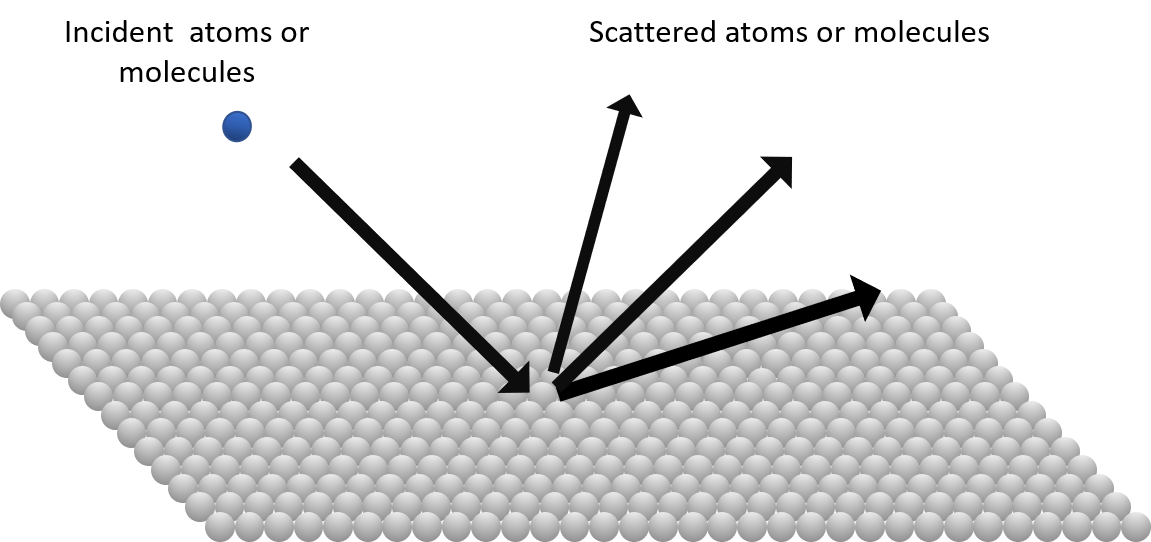
Figure 1. The principles of a scattering experiment. The incident particles are prepared and characterized with respect to their energies (translational and internal) and angles of incidence. Molecular beam techniques and optical excitations are major preparative tools. The scattered particles are analyzed with respect to their energies (translational and internal) and angular distributions. Mass spectrometry and optical (laser) techniques are major analytical tools.
The overall ambition in dynamics studies is thus to understand, at an atomic level, how things happen and evolve at the surface. Typical questions asked are: “How can a molecule that bounces towards a surface lose enough energy to stick to it?", "Which are the detailed steps involved in desorption of atoms and molecules from surfaces?”, and “What causes or prevents dissociation of simple molecules like H2, CO, O2 on different surfaces?”. “How do two different atoms/molecules react on a surface to create a new molecule, as in a catalytic reaction?” Some of the elementary processes in focus here are shown in Fig. 2, and for a specific system in Fig. 3. Adsorption, desorption and diffusion events of simple atoms and molecules, like noble gas atoms and H2, CO, O2, NO, became popular model systems for studies of the collision dynamics and associated energy and charge transfer events.
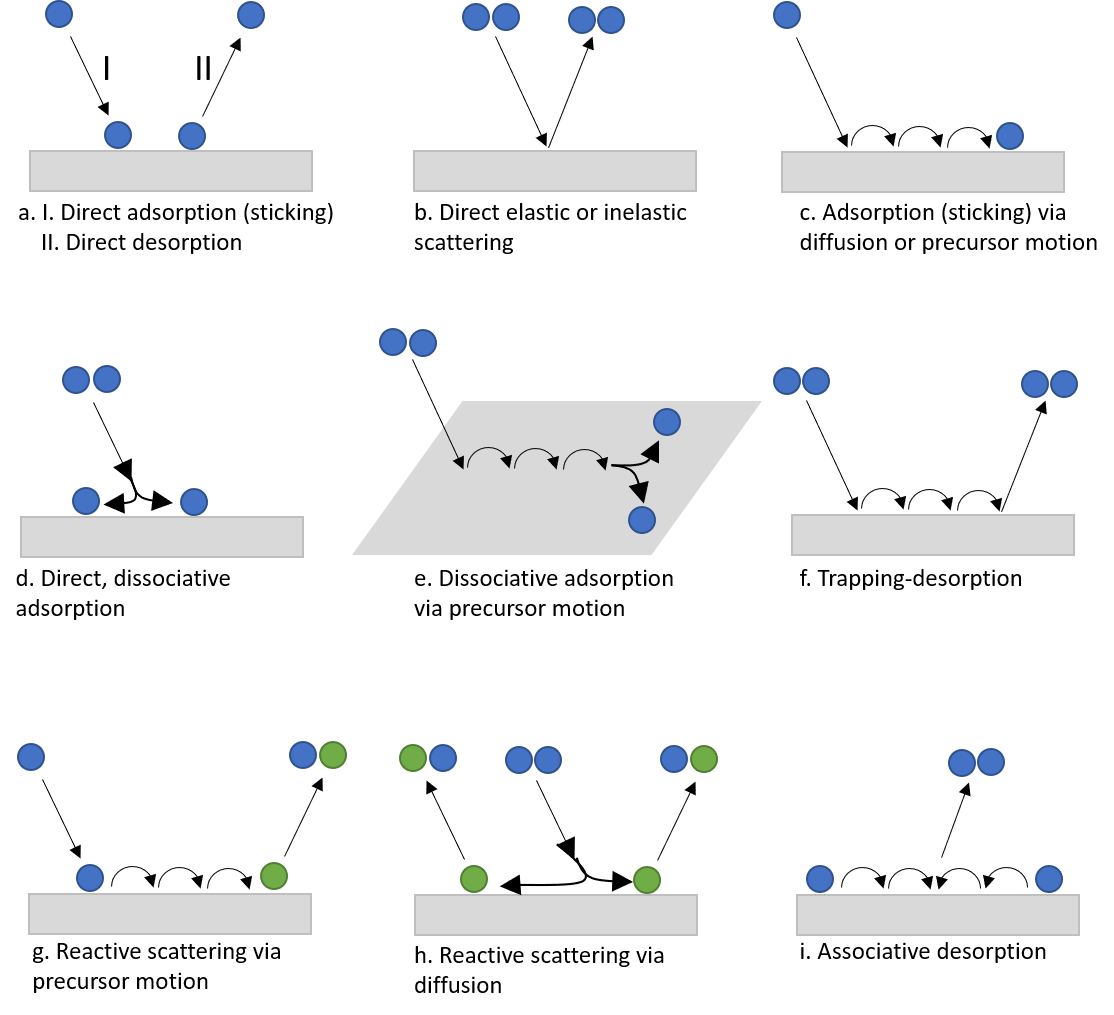
Figure 2. Examples of gas-surface interaction events referred to in the text.
Qualitatively what we want to understand in gas surface dynamics are (Fig. 2) how molecules or atoms from the gas phase interact with surfaces. For example, how do they land on the surface (sticking, adsorption, chemisorption), exchange energy with it, and then (sometimes) leave it?
As an illustration of what dynamics address and contain, we imagine the space-time path of a CO molecule approaching a partially oxygen-covered catalyst surface like Pt (Fig. 3). First, it lands on the surface (sticking) by getting rid of some of its excess energy, then it diffuses over the surface, i.e., jumps between different adsorption sites, and eventually reacts with an adsorbed oxygen atom and thereby forms a CO2 molecule.
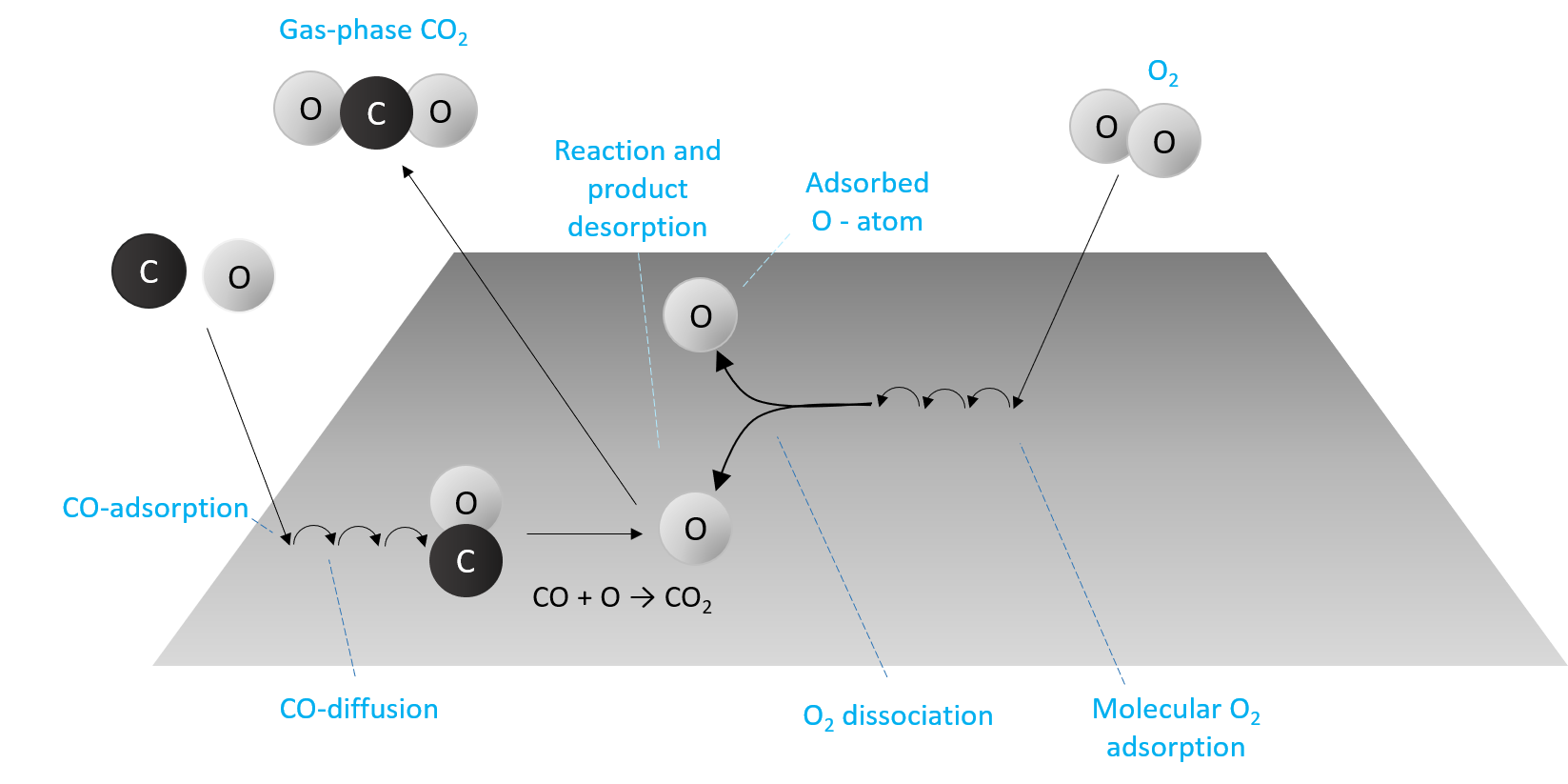
Figure 3. Illustration of the system CO + O2 → CO2 + O
A useful concept and quantity in the discussion of dynamics is the potential energy surface (PES), Fig. 4. A point on a PES, e.g., for an atom on a real surface, corresponds to the total energy of the whole system at that point. In real space, the point is defined through all the relevant coordinates in the system and the total energy of the system at that point. For an atom, the coordinates are the atom’s distance from the surface and the x-y coordinates parallel to the surface. This gives a four-dimensional space for the atom-surface PES (
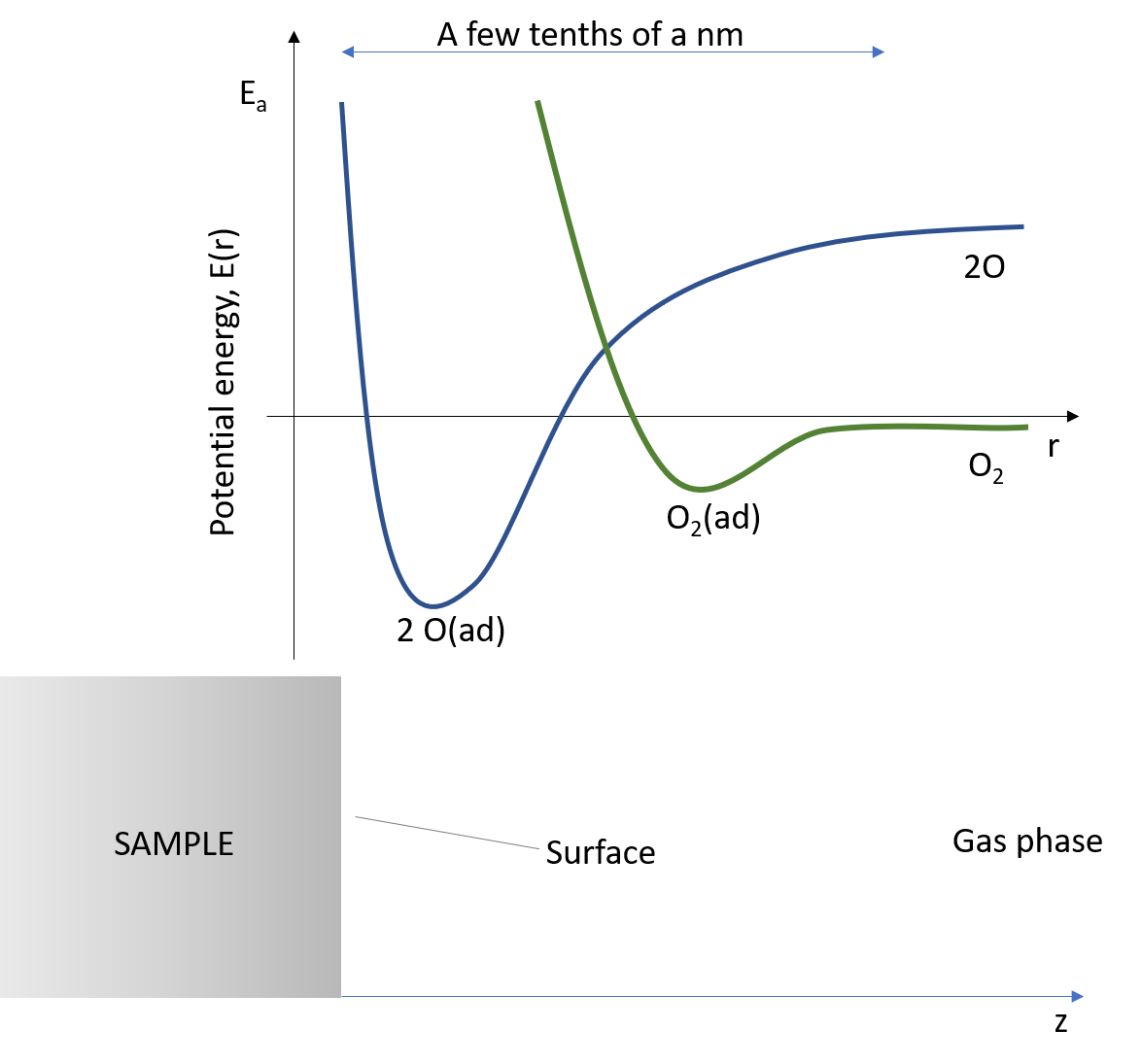
Figure 4. A one-dimensional cut of the 6-dimensional PES for a system like O2 on Pt, where there exists a molecular adsorption well (minimum) in the entrance channel for the molecule to the surface, and then an activation barrier for dissociation of the O2 molecule and final formation of two chemisorbed O atoms. This way of drawing the 1 D cut of the 6 D PES means that the coordinate r is rather complex – it is not just the vertical distance to the surface, z, but runs through
When addressing dynamic events on surfaces, the static ground state properties, i.e., the lowest energy state, is insufficient as a platform for the description. The static ground state is
The timescale of a simple molecule scattering event, with no trapping or sticking, but just a single collision, where the molecule bounces back into a vacuum, is typically 10-12 – 10-13 seconds. This is the same as one vibrational period of a molecule in free
If a molecule has been thermally equilibrated with the surface, it can desorb due to thermal fluctuations, if the combination of temperature and binding energy to the surface are in the right range. The timescale for the desorption event, or surface "lifetime" of the molecule, can be estimated using the Arrhenius approximation (see below), where the time constant, τd, for
For classification of gas surface interaction/scattering events one frequently uses the three classes I) elastic, II) inelastic and III) reactive scattering. In the former two, all chemical bonds are preserved, and new ones are not formed, while bond-breaking and bond-formation characterize class iii). The term “scattering” is used because in the more detailed studies one always uses molecular beams, where the beam atoms or molecules scatter against a single crystal surface (Fig. 1).
A “cleanest” example of elastic scattering is when He atoms scatter against a single crystal surface. A unidirectional and monoenergetic beam of He atoms scatters predominantly without energy deposition, or energy pick-up, to/from the surface. One branch of studies even focuses on diffraction of He atoms to extract structural information about the scattering surface (
In inelastic scattering, energy is deposited into the surface and/or picked up from it. Possible energy conversion events are shown in Fig. 5. For atoms, a typical example is heavier noble atoms than He, e.g., Ar, and especially Xe, where kinetic (translational, T) energy of the incident atoms can be transferred to, or picked up from, the phonon bath (vibrations, V) of the surface atoms. If the incident particle is a molecule, additional energy conversion/dissipation events are possible like rotational (R) and vibrational (V) and even electronic (E) excitations. In the latter cases, the molecules in the molecular beam are prepared with specific vibrational, rotational or electronic excitations.
On the detection side, after scattering, one measures the changes in these vibrational-rotational-electronic quantum numbers by laser spectroscopy and translational energy changes by pulsed/chopped beam techniques. Large sets of data are obtained by varying the initial vibrational-rotational quantum numbers, translational energy and angles of incidence. With theoretical (molecular dynamics) tools, one then tries to make models of the surface PES and of how all the scattering events (elastic and inelastic) take place.
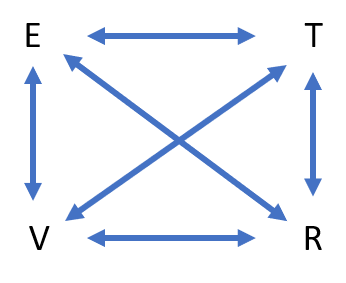
Figure 5. Possible energy conversion events in molecule-surface scattering events.
T:
V:
R:
E: Electronic excitations in the incident or scattered atoms/molecules and/or in the surface region (e.g., electron-hole pairs).
Especially in connection with elastic and inelastic scattering the concept “surface corrugation” is often used. This concept describes how ‘corrugated’ the energy landscape of the PES under study is. As we learned above, a point on the PES is the total energy of an atom/molecule at a specific point in real space. Corrugation refers to how much this energy varies as we move over the PES, or more correctly how much that energy varies
In reactive events chemical bonds are broken and/or new ones are formed. One of the most widely explored sub-class here is catalytic reactions of the formal type A + B → C where A, B are the original reactants and C the reaction product. The catalyst surface enhances the reaction dramatically, without being changed itself. But there are also other types of reactive events, e.g., reactive etching where A species from the gas phase may react with a surface atom, B, to form a gas phase product C, thereby removing surface material (often chemically and/or spatially selective).
Experimentally reactive scattering can be performed with one or two molecular beams (MBs). In the former case one typically first deposits some predetermined coverage of atoms/molecules, say reactant A, on the surface. Then one directs molecular beam molecules of B to the surface and detects the product molecules AB for varying incident MB parameters and for different surface temperatures. One may also detect the velocity and angular distribution of product molecules AB.
The experiment may also be reversed, i.e., by exchanging A for B. With two molecular beams, there are more opportunities for variability in the experiment. One example is that a steady state can be established with two beams, where the surface coverage stays constant, but the reaction goes on continuously. This is often made at different ratios of the A/B fluxes. One may furthermore catch reaction events involving transient states of reactants on the surface, like molecules in precursor motion.
Dry etching reactions constitute a sub-class of reactive scattering. Dry etching is a method used extensively in semiconductor surface processing in combination with photolithography for microelectronics. For example, a silicon surface may be dry etched with fluorine-containing molecules. A typical example is XeF2 etching of the Si surface by forming SiF4 which desorbs from the surface. The net result is a removal of one Si atom from the surface from two incident XeF2 molecules. In practice, the etching is performed in a plasma etcher, but MB experiments can help elucidate the reaction mechanisms.
Above I have outlined the essence of gas-surface dynamics. There are some additional concepts that are common in dynamics and which I briefly define and comment on below. It is also appropriate to articulate a few special cases like ‘sticking’ and thermal desorption. Let’s begin with ‘sticking’.
The ‘sticking’ of molecules and atoms on surfaces is a central concept in surface reactions. Sticking is essentially synonymous with “adsorption” as a process and leads to adsorbed/chemisorbed molecules. It connects kinetics and dynamics via the adsorption rate constant ka. The kinetic equation (no desorption) for first-order adsorption (commonly seen for adsorbing atoms) is
dθ/
where θ(t) is the surface coverage. ka is the product of the gas impingement rate (gas pressure) and the sticking probability, s0, on the clean surface. ka contains via s0 (in an embedded form) all the atomistic details of the scattering event when a molecule hits the surface.
The simple solution to the Eq. 1 is
θ(t) = 1- e-t/τ (2)
and τ=1/ka
The solution describes how an initially clean surface, exposed to a constant flux of gas species, fills up to saturation in the same functional way as a capacitor is charged with a constant voltage. Higher gas flux and higher values of s0 shorten the time scale τ to fill up the surface with adsorbates. Typically, the time constant for filling the surface is of order a few seconds at 10-6 torr pressure and s0=1. First order adsorption is often called monoatomic adsorption but can also apply for simple molecular adsorption. If the type of adsorption is instead second order, common for dissociative adsorption of a diatomic molecule, we have dθ/
Sticking coefficient measurements have been subject to many studies over the years. Methods range from surface spectroscopic or desorption measurements of the coverage θ(t) for different gas doses to the
Numbers lower than unity are indicative of the existence of an activation barrier for adsorption. In such cases it may be elucidating to measure s0 at different incident energies of the molecule; an activation barrier may prevent slow molecules from sticking but not sufficiently fast ones. An activation barrier may also be overcome by internal vibrational or rotational energy of an incident molecule, due to concerted adsorption and energy conversion events (Fig. 5).
A smaller than unity sticking coefficient can, however, also be seen without an activation barrier due to inefficient energy transfer. This happens when the molecule/atom does not dissipate energy sufficiently fast in one single scattering event, to be trapped by the surface and therefore returns to the gas phase.
A sub-class of experiments in this area, not requiring molecular beams, but just a gas inlet to adsorb molecules on a
dθ/
where
kd=ν exp(-Ed/kT) (4)
Ed is the desorption energy, which for non-activated adsorption is essentially equal to the molecule’s binding energy to the surface. ν is the so-called preexponential factor, which in simple absolute rate theory has a value of order 1013.The applied temperature rise is an experimental parameter and enters as T(t) = ct, where c is the heating rate, and where
For diatomic molecules that dissociate upon adsorption, like O2 on many metals, the desorption is frequently found to be 2nd order in the coverage, i.e., dθ/
A molecule outside a surface, to which it eventually binds, constitutes an excited state of the system. When a molecule-surface bond is formed, a lower energy state is formed, and in the process, the initial higher energy of the system must in some way be dissipated.
The candidates for energy dissipation are many (Fig. 5). A strong candidate is the phonons (lattice vibrations) of the solid surface. As, e.g., a heavy and energetic Xe atom hits a surface, a major energy dissipation channel is phonons. Since the energies involved are typically much larger than that of single
Another surface candidate is electronic excitations, i.e., electron-hole pairs. It is less common, but increasingly more important, as the mass ratio of incident particles and surface atoms get smaller, making phonon excitations less effective, e.g., hydrogen molecules on transition metals.
Yet another example (reactive interaction) is when electronegative molecules, like Cl2, hit an electropositive (low work function) surface. Then, electron transfer can occur from the surface to the (electron affinity level of an) approaching Cl2 molecule and lead to electron-hole pair creation as an energy dissipation channel (see more about this case below). The lifetime of electron-hole pair excitations is extremely short for metals, 10-14 – 10-15 s. Phonon lifetimes are one to two orders of magnitude longer.
But the surface excitations (phonons and electron-hole pairs) are only one set of energy dissipation channels (Fig. 5) associated with the surface. For molecules, there are also the internal molecular excitations that can pick up or give away energy, namely rotations and vibrations. In reality, a full scattering/sticking process may involve many of these elementary excitations and energy conversion events (the arrows in Fig. 5), simultaneously or sequentially. The lifetime of molecular vibrations at surfaces are in the range from tens of picoseconds to fractions of picoseconds - orders of magnitude shorter than in the gas phase, due to the efficient channeling of energy into phonons or electron-hole pairs.
It is also possible that a molecule with excess kinetic, i.e., translational energy, collides with the surface and converts translational energy to internal, rotational or vibrational energy of the same molecule, which then bounces off the surface after a translational energy-to-internal energy conversion. Fast incident molecules can thus scatter on the surface back into a vacuum or gas phase, with much higher internal vibrational-rotational excitation than in the incident molecules (and lower translational energy).
The discussion above can also be applied to the reverse process to adsorption, i.e., desorption. This step, when a molecule or atom leaves the surface, requires energy pick up from the surface (from its heat-bath of phonons,
Another aspect of the scattering and energy dissipation, that has generated much interest and work, is whether the energy dissipation in e.g., an adsorption event is adiabatic or non-adiabatic, which in turn affects on which PES the adsorbing particle moves.
If energy dissipation happens gradually, like in a friction process, and the energy is continuously dissipated as very low energy quanta, e.g., as phonons, i. e. heat, we call the energy dissipation and adsorption process adiabatic. However, in many cases, the system does not dissipate energy adiabatically but in larger, sudden (quantum) energy release events, like a photon, or an electron or a cascade of
The scattering processes mentioned above are preferably studied with molecular beams. This allows well-defined translational and internal energies of the molecules in the beam. The outcome of such collision and energy conversion/dissipation events are studied by analyzing the scattered molecules with respect to angular distributions, translational energy and internal energy, and also by measuring the fraction (if there is one) of molecules stuck on the surface. Of course, also the parameters of the incident molecules (angle of incidence, translational and internal energies, etc.), are varied. The collective name of this area became gas-surface dynamics.
The scattering events take place in a multi-coordinate space. For example, to uniquely define the scattering of a two-atom molecule against a single crystal surface several aspects must be defined; (i) the direction of the molecule i. e. the azimuthal and polar angle of its trajectory to the surface, (ii) where does the molecule hit the surface in the surface unit cell (e.g., on top of a surface atom, or in the hollow between three or four surface atoms), and (iii) what is the rotational orientation of the molecule, and what is the vibrational phase. To develop a theory for the scattering event, the calculations need to include all these coordinates. Since all of them cannot be measured and some are averaged over all possible values in the experiment like the rotational orientation and vibrational phase, a challenge is to use all experimentally accessible information about the incident beam and the scattered molecules to learn about the scattering details and participating the PESs.
When a strongly electronegative molecule like Cl2 approaches a metal surface with low work-function like potassium, the electron affinity level of Cl2 shifts downwards in energy due to so-called image interaction. As a result, an electron can jump or tunnel from the potassium surface to the Cl2 molecule, forming a transient Cl2- molecule (the affinity level is the lowest unoccupied electron level in the Cl2 molecule, i.e., where the weakest bound electron of a Cl2- sits.) As a result, the Cl2 molecule gets stuck to the surface and dissociates to form two KCl bonds on the surface. The timescale for the involved processes
If the incident particle is instead very electropositive and the surface has a high work function, like K or Na incident on Pt or W, the reverse process occurs. Electrons tunnel from the alkali atom’s valence orbital into the valence band of the
In the context of adsorption/sticking, precursors and precursor motion are frequently appearing concepts. This usually refers to a motion of a molecule/atom just after it has struck the surface, but before it has dissipated its excess energy and come to rest and equilibrated with the temperature of the surface (Fig. 2c). The motion on the surface is driven by the adsorption energy and occurs before that energy has been fully dissipated.
There are several possible mechanisms for precursor motion. The molecule may in the first encounter convert some of its momentum normal to the surface into parallel momentum (elastically) or just dissipate that part of its energy (inelastically), resulting in a ‘hot’ precursor moving along the surface with higher than thermal energy, but trapped by the surface interaction. During the parallel motion, which occurs on the corresponding PES, the excess energy may be successively dissipated until the molecule is trapped in some local or absolute minimum on the PES and just becomes an adsorbed and thermally equilibrated molecule. The motion is similar to diffusion, but the latter is purely thermal in nature. A hot precursor may visit parts of the PESs not accessible via thermal diffusion.
An alternative outcome of the “hot” precursor motion on the surface is that the precursor regains energy and/or vertical momentum and scatters back to the surface. This is sometimes referred to as a trapping-desorption channel (Fig. 2f).
Another type of
When a surface with adsorbed atoms/molecules is irradiated with electrons of a few or a few tens of eV, i.e., a little more than the surface chemical bond strengths, it may lead to a special type of desorption, called photo-induced (PID) or electron stimulated (ESD) desorption. The mechanisms involved in such events are almost always electronic excitations, either in the adsorbed molecule itself or, more common, in the surface or in the molecule-surface bond.
A common case is excitation of an electron-hole pair in the valence band of the surface. Typically, the initially excited hot electron moves to the adsorbate and into an antibonding orbital of the adsorbate – surface complex, which in turn causes the adsorbate to be repelled from the surface into a vacuum. Alternatively, it is instead the excited hole state that moves to the adsorbate and empties a bonding orbital, thereby causing desorption. Similar events may lead to dissociation of adsorbed molecules, which in turn may cause a surface
One of the most studied model reactions in the reactive scattering category is CO reacting with an oxygen atom, the latter derived from a dissociated O2 molecule (Fig. 3) or NO2
This simple scenario (previous paragraph) hides some essential details of the reaction, which are discovered if one runs the experiment at a much lower temperature and in a slightly different way. When CO and O2 are co-adsorbed on Pt at sufficiently low temperature, typically well below 150 K, both molecules stay intact and non-dissociated on the surface, and they do not react. If the surface temperature is then raised monotonically with co-adsorbed CO and O2, one suddenly sees CO2 molecules leaving the surface already around 150 K, indicating reactions between the absorbed CO and oxygen. However, only a fraction of the CO molecules reacts at this temperature, and a new desorption flux of CO2 is seen when the temperature reaches around 330 K. The latter is caused by the same reaction as was just discussed above between adsorbed CO and O atoms.
The actual events that occur are that the low-temperature formation of CO2 is caused transiently by dissociating O2 molecules (thermally activated dissociation). Some of the O atom dissociation products react with neighboring CO molecules and some just form adsorbed oxygen atoms as in Fig. 3. The latter cannot react with remaining CO molecules on the surface, because of a rather high activation barrier, until the temperature reaches around 300-350 K. The latter is the ‘normal’ CO oxidation reaction on Pt, with which this paragraph began, and which has been studied extensively since the days of Langmuir. The former reaction occurring at around 150 K is in a way more exotic, but mechanistically interesting, and reveals that there is a stable adsorbed molecular O2 state that is permanently occupied at sufficiently low T and transiently at high T. This state has been spectroscopically verified.
If in the latter experiment, with co-adsorbed O2 and CO molecules, at < 150 K, where they don’t react, the temperature is not raised, but instead a flux of near UV photons is sent to the surface, CO2 molecules are also formed, without any T increase. The mechanism of this reaction is first a photoexcitation of electron-hole pairs in the Pt surface, followed by (hot) electron transfer to an antibonding orbital of the adsorbed O2 molecule, forming a transient O2-, thereby inducing its dissociation to O atoms. The latter helps to get over the activation barrier in Fig. 4. Some of these O atoms bounce into and react with neighboring CO molecules, and form CO2, in much the same way as in the TPR experiment.
In PES language, this experiment shows that there exists a ground state PES for O atoms with a deep minimum corresponding to chemisorbed O atoms, and an excited state PES for O2, with a shallow minimum for O2. It also shows that there is a local minimum for an adsorbed O2 molecule with an activation barrier towards dissociation
My ambition with this post has been to present an overview of the field called gas surface dynamics, its general concepts and phenomena and what this field is all about. I have covered scientific questions and types of studies embraced by this area. Detailed descriptions of specific model systems are beyond the scope of this post. For example, the descriptions of adatom or
As also mentioned in post 1 on static systems, I want to emphasize the important role played by theory, both for interpretation of experiments, for developing concepts and understanding and for planning experiments. The theory aspect will be discussed in a later post, as will the relevant and extensive topic of experimental methods be.
Finally, as was indicated in the introductory part of this post, dynamics of molecule-surface interactions are very far from atomistic detail for more complex systems. Let’s take as an example protein-surface interactions. Even if we choose the simplest possible, yet relevant surface, like graphite or gold, and the simplest possible protein, we are still very far from an atomistic description. The latter would require describing a very complex and multidimensional PES on a time scale from picoseconds to seconds or even minutes, keeping track of all the 100ds or more atoms in the protein and how they (re)orient, bind to the surface, break or form bonds etc., creating a full molecular-dynamics ‘movie’ of how the protein binds, orients, unfolds, (partially) denatures etc. Although we are very far from such descriptions it is the direction of future studies.

Bengt Kasemo was one of the co-founders of Q-Sense AB in 1996 and was on the Board of Directors of Q-Sense and Biolin Scientific AB for several years. Professor Kasemo's research projects focused on surface science, bio-interfaces, biomaterials, catalysis, nanotechnology, sensors and scientific instruments, nanotoxicology and nanosafety, as well as sustainable energy.

